Surgery versus non-operative treatment for ER-stress unstable Weber-B unimalleolar fractures: a study protocol for a prospective randomized non-inferiority (Super-Fin) trial
Roughly two-thirds of ankle fractures are unimalleolar injuries, the Weber B-type fibula fracture being by far the most common type. Depending on the trauma and the accompanying soft-tissue injury, these fractures are either stable or unstable. Current clinical practice guidelines recommend surgical treatment for unstable Weber B-type fibula fractures. An ongoing randomized, parallel group, non-inferiority trial comparing surgery and non-operative treatment for unstable Weber B-type ankle fractures with allocation ratio 1:1. The rationale for non-inferiority design is as follows: By being able to prove non-inferiority of non-operative treatment, we would be able to avoid complications related to surgery. However, the primary concern related to non-operative treatment is increased risks of ankle mortise incongruency, leading to secondary surgery, early post-traumatic osteoarthritis and poor function. After providing informed consent, 126 patients aged 16 years or older with an unimalleolar Weber B-type unstable fibula fracture were randomly assigned to surgery (open reduction and internal fixation) or non-operative treatment (6-week cast immobilization). We have completed the patient enrolment and are currently in the final stages of the 2-year follow-up. The primary, non-inferiority outcome is the Olerud-Molander Ankle Score (OMAS) at 2 years (primary time point). The predefined non-inferiority margin is set at 8 OMAS points. Secondary outcomes include the Foot and Ankle Score, a 100 mm Visual Analogue Scale for function and pain, the RAND-36-Item Health Survey for health-related quality-of-life, the range-of-motion of the injured ankle, malunion (ankle joint incongruity) and fracture union. Treatment-related complications and harms; symptomatic non-unions, loss of congruity of the ankle joint, reoperations and wound infections will also be recorded. We hypothesize that non-operative treatment yields non-inferior functional outcome to surgery, the current standard treatment, with no increased risk of harms.
Introduction
Background and rationale
Seventy per cent of ankle fractures are unimalleolar injuries, the Weber B-type of fibula fracture being by far the most common type.1–8 In this particular fracture type, the ankle mortise is either stable or unstable depending on accompanying soft tissue injury.5–15 External-rotation (ER) stress testing is the most reliable means to assess the stability of the ankle mortise.8 10–12 Ankle mortise stability has fundamental clinical relevance, as it dictates the subsequent treatment strategy.5–16 If left untreated, an unstable ankle mortise may lead to compromised fracture healing, increased risk of post-traumatic osteoarthritis, and subsequently, poor functional outcome.5–7 10–14 Therefore, existing literature quite unanimously recommends surgery for unstable Weber-B fibular fractures.5–7 10 12–14 The gold-standard surgical treatment for these fractures is open reduction and internal fixation.6 7 16 17 The most common complication following operative treatment of ankle fracture is wound infection, the incidence ranging from 6.1% to 10% in unselected patient materials.18–21
To date, there is only one published randomized trial comparing surgery and non-operative treatment in patients with an unstable unimalleolar fibula fracture.22 At the 1-year follow-up, the functional outcomes of the two groups were equivalent but in patients treated initially non-operatively, the overall incidence of compromised fracture healing was 40% (8 patients with fracture displacement and 8 with delayed or non-union) while 10 of the 41 patients randomized to surgery (24%) had treatment related complication or harm: 6 patients with post-operative wound infection, of which one needing revision surgery and additional 4 patients with symptomatic hardware requiring removal .22
Super-Fin (SF) is a prospective randomized non-inferiority trial designed to compare surgery to non-operative treatment in patients with an ER-stress positive unimalleolar ankle fracture. The primary, non-inferiority, intention-to-treat outcome is the Olerud-Molander Ankle Score (OMAS)23 at 24 months. Our hypothesis is that 6-week cast immobilization yields non-inferior functional outcome to surgery, with no excess incidence of harms by potentially avoiding complications related to surgery. Non-inferiority of the non-operative treatment with respect to surgery is of interest as non-operative treatment has some other benefits,24 such as being less burdensome to the patients and the healthcare system. We consider non-inferiority proven if ankle function in the non-operative group, as determined by OMAS, is within the predefined non-inferiority margin of the surgery group and there is no significantly increased risk of harms. Our predefined non-inferiority margin for the primary outcome at the primary assessment time point is set at eight points.
Methods and analysis
Study design and rationale for non-inferiority
This paper describes a research protocol for the SF-Trial, an ongoing, prospectively registered, randomized controlled non-inferiority trial comparing surgery and non-operative treatment for unstable Weber B -type ankle fractures. The protocol was developed in accordance with Standard Protocol Items: Recommendations for Interventional Trials, and Template for Intervention Description and Replication statements.25 26
The rationale for non-inferiority design is as follows: By avoiding surgery, we would be able to avoid characteristic harms related to surgery (the risks of anesthesia, wound healing problems/infection, and the need of symptomatic hardware removal). The primary concern related to non-operative treatment is increased risk of malunion (incongruent ankle mortise), predisposing to early post-traumatic osteoarthritis and poor function. Our secondary outcomes are thus geared at assessing ankle mortise congruity, early signs of ankle joint osteoarthritis (at mid- to long-term outcome: 5- to 10-year timepoint) and the recovery of the ankle range-of-motion (ROM).
Study setting
This ongoing trial is conducted at the Oulu University Hospital, which has a catchment area of approximately 250 000 inhabitants for fractures. Before the launch of the trial, we estimated that roughly 40–50 patients with an unstable unimalleolar Weber B-type fibula fracture, suitable for the study, would be treated at our hospital annually. Patient screening began in January 2013 and we reached our recruitment target (126 patients) in March 2019. Due to COVID-19-related delays, we estimate to complete the follow-up in June 2021.
Eligibility criteria and primary assessment
Inclusion and exclusion criteria are listed in box 1.
Box 1
Inclusion and exclusion criteria
Inclusion criteria
Skeletally mature patients (16 years or older) men or women.
Isolated Weber B-type fibula fracture and no widening of the ankle mortise on the static ankle radiographs.
Medial clear space (MCS) < 4 mm and ≤ 1 mm wider than the superior clear space.
Unstable ankle mortise at the ER Stress test.
MCS ≥5 mm as measured between the lateral border of the medial malleolus and the medial border of the talus at the level of the talar dome.
Patients able to walk unaided before the current trauma.
The enrolment and operative treatment within 7 days from injury.
Provision of informed consent.
Exclusion criteria
Ankle fracture-dislocation.
Previous ankle fracture or deltoid ligament injury or other significant fracture in the ankle/foot area.
Bilateral ankle fracture.
Concomitant tibial fracture.
Pathological fracture.
Diabetic or other neuropathy.
Inadequate co-operation.
Inability to speak, understand and read in the language of the clinical site (history of alcoholism, drug abuse, psychological or psychiatric problems that are likely to invalidate informed consent).
Permanent residence outside the catchment area of the hospital.
Open fracture.
Patient declined to participate.
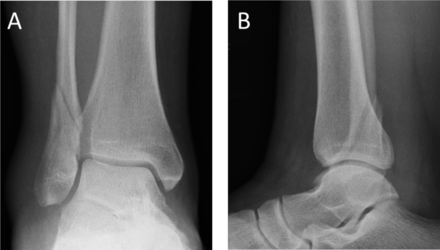
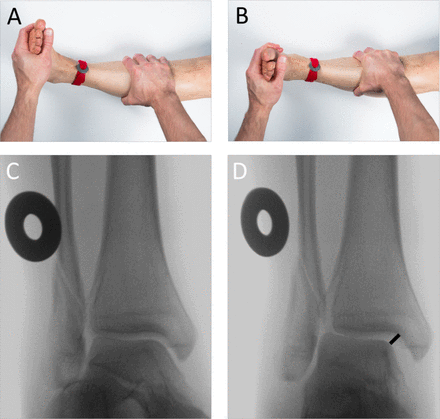
All skeletally mature patients (16 years or older) with an isolated (ie, no other osseous injury) Weber B-type fibular fracture with congruent ankle mortise (figure 1) were assessed for study eligibility. Ankle mortise was considered congruent when the medial clear space (MCS) was <4 mm and ≤1 mm wider than the superior clear space at the mortise-view in standard non-weight-bearing radiographs with ankle in neutral dorsiflexion. All these patients underwent an ER stress test under fluoroscopy (ER-stress test, figure 2A,B) to assess stability of the ankle mortise.10–12 The fracture was considered unstable when the MCS was ≥5 mm as measured between the lateral border of the medial malleolus and the medial border of the talus at the level of the talar dome (figure 2C,D).10–12 The fluoroscopy radiographs were calibrated using a 30 mm disk placed and fixed with tape to the skin of the patient ankle just above the upper ankle joint line. The accuracy of the measurements is 1 mm.
External rotation (ER) stress test. To obtain an approximation of the true mortise view, the leg is stabilized in an approximately 10°–15° of internal rotation, with the ankle in neutral dorsiflexion (A).10–12 A fluoroscopy scan is first obtained to ensure correct positioning (C). An ER force of approximately 8 –10 lb/3.6–4.5 kg is then applied to the forefoot before repeating the scan (B, D). A 30 mm calibration disk is used to calibrate the radiographs (A), with measurements made to an accuracy of 1 mm. The fracture is considered to be unstable when the medial clear space, measured between the lateral border of the medial malleolus and the medial border of the talus at the level of the talar dome, is ≥5 mm (D).
Informed consent
After ER-stress testing, all eligible patients were introduced to the trial, provided detailed written information, and then asked to participate by giving written informed consent. Patients were informed that they can withdraw from the study at any time, without affecting the course of their treatment, in accordance with the latest version of the Declaration of Helsinki 2013.27
Baseline data
After consenting, the following baseline data was gathered: Birth date, sex, fracture type, injury date, clinical findings on the medial side of the ankle, pain at the ER-stress test (Numeric Rating Scale) and MCS, mm at the ER-stress test.
Allocation
Sequence generation and concealment
Patients were randomly allocated (1:1 ratio) to 6 week cast (non-operative treatment) or surgery using sealed and consecutively numbered, opaque envelopes. A biostatistician who was not involved in the clinical care of the patients prepared the envelopes using a computerized random number generator. The envelopes were kept in a secure place only known by trial personnel. To minimize the risk of predicting the treatment assignment of the next eligible patient (to ensure concealment), randomization was performed with random permuted blocks (block size known only by the statistician).
Stratification
No stratification was used.
Implementation of randomization
Orthopedic surgeon on-call randomized patients individually by opening the next available sequentially numbered (from 1 to 126) envelope containing the group assignment. These surgeons were not involved in any further treatment of the trial patients.
Blinding
Blinding of participants and outcome assessors was not possible because of the nature of the interventions. However, the OMAS score (primary outcome) and most of our other secondary outcomes are patient-reported outcomes.
Interventions
Non-operative treatment
For patients allocated to non-operative treatment, the injured ankle was placed in a standard, padded below the knee synthetic cast by a trained plaster technician. The ankle joint was immobilized at 90° angle (figure 3). Participants received guidance from a physiotherapist on walking with crutches. Immediate partial weigh-tbearing (approximately 15–20 kg) was permitted after the application of the cast and patients were instructed to start full weight-bearing (as tolerated) at 4 weeks after the injury.
A standard, padded below the knee synthetic cast made by a trained plaster technician. Cast is applied from the tuberosity of the tibia to the base of the toes and is lined and padded. The cast is applied with the ankle joint placed at 90° angle (neutral dorsiflexion).
Surgical treatment
For patients allocated to surgery, either a backslab or a cast was placed at the emergency department: A backslab if soft-tissue condition/injury allowed early surgery (positive skin wrinkle sign, no blisters) and a contemporary cast if severe soft-tissue injury (swelling, negative wrinkle sign and/or blisters) existed. For those with severe soft-tissue injury, surgery was carried out as soon as the swelling subsided.
At the day of surgery, all patients received standard preoperative antibiotics (cefuroxime 1.5 g–3.0 g or clindamycin 300 mg–600 mg depending on body mass index) before skin incision. Surgery was performed according to standard principles for ankle fracture fixation: Direct reduction and lag screw fixation of the fibular fracture when possible, and fibular stabilization using a neutralization or antiglide 1/3 semitubular plate. The use of tourniquet was left at the discretion of the operating surgeon. All surgeries were performed during the office hours by experienced orthopedic trauma surgeons or by orthopedic residents under the direct supervision of an orthopedic trauma surgeon. Wounds were closed in two layers with the skin closed either using stitches or staples. Unless major swelling or wound bleeding, all operated ankles were placed on a below-the-knee cast identical to the non-operative group on the first post-operative day. The guidance on walking with crutches and the instructions on weight-bearing were identical to the non-operative group.
All trial patients received written and verbal instructions on to how to cope with the ankle fracture and the cast. We did not perform a formal registration of problems related to ill-fitting or compliance with the casts but asked all trial patients to contact the study hospital if they experienced any issues with the cast. In such events, a new cast was applied. At the 2-week follow-up, the cast was removed, wound inspected and stitches or staples removed (surgery group), and a new cast applied.
The duration of initial sick leave was defined by the surgeon-on-call in accordance with the requirements of the patient’s work.
At each follow-up visit from 6 weeks onwards, patients received instructions from a study physiotherapist on ankle rehabilitation.
Follow-up appointments and timetable for follow-ups
The outcomes used in this study and the timetable for follow-up assessments are summarized in table 1.
Table 1
|
Schedule of follow-up and outcome assessments
Clinical follow-up visits are scheduled at 2, 6 and 12 weeks, and at 2 years after randomization. The visits include a clinical examination and radiography (mortise and lateral projections) of the injured ankle. Prior to the final 2-year follow-up, the participants were mailed the study questionnaires and asked to independently complete questionnaires assessing ankle functional outcome, pain and quality of life.
Outcomes
The outcome measures are similar to those described in our previously published study on non-operative treatment of ankle fractures.28
Primary non-inferiority outcome
The primary outcome measure is the OMAS (scale 0–100, with higher scores indicating better outcomes and fewer symptoms), a validated, condition specific, patient-reported measure of ankle fracture symptoms.23 29 OMAS is an ordinal scale, but with 21 classes (scale 0–100, at intervals of 5 points), it is close to numerical continuum and is handled as such in the statistical analysis. The primary time point was at 2 years, as predefined in the trial registration.
Secondary outcomes
The secondary outcome measures are the Foot and Ankle Outcome Score (FAOS, five subscales from 0 to 100, with higher scores indicating better function),30 a 100 mm Visual Analogue Scale (VAS) for function and pain (range 0–100, with higher scores indicating more severe pain or dysfunction),31 the RAND-36-item health survey for health-related quality of life (RAND-36, eight subscales from 0 to 100, with higher scores indicating better quality of life),32 ROM of the injured ankle measured using a goniometer,33 34 malunion (ankle joint incongruity) determined from radiographs (yes or no), and fracture union (assessed at 2 years). We will consider fracture union to be complete when the fracture line has disappeared, and non-union present when the fracture line is still visible. Two experienced orthopedic surgeons with no access to clinical data or patient reports will analyze the radiographs. Except for malunion (assessed at 2, 6 and 12 weeks and at 2 years) all outcomes are assessed at 2 years, the study primary time point.
Radiographs
Ankle joint congruity of the injured ankle is assessed using both mortise and lateral X-ray projections at every follow-up visit, while fracture union will be assessed at the 2-year follow-up only. The mortise view is carried out with the leg internally rotated 15°–20° so that the X-ray beam is perpendicular to the intermalleolar line. This view permits examination of the articular space (clear space). Ankle mortise is defined normal (congruent) when MCS is <4 mm and ≤1 mm wider than the superior clear space at the mortise view.
ROM of the injured ankle
ROM is measured using a goniometer at the 2-year follow-up visit by a physiotherapist aware of the treatment groups. Maximum dorsiflexion is determined with the patient standing on the injured ankle on a 30 cm high investigation table, asked to lean as far forward as possible with his/her heel remaining on the table. Plantarflexion is determined with the patient sitting on an examination plane and then asked to plantar flex his/her injured ankle. The angle is then measured between the fifth metatarsal and fibula. Measurements are made to an accuracy of 5°.33 34
Safety considerations
Expected complications or harms related to study treatments, which included loss of congruity of the ankle joint, venous thromboembolism, wound infection, implant failure, fracture non-union and re-fracture, are recorded as adverse events. In addition, we will record unexpected adverse events. At each follow-up visit we will query about harms, and participants are asked to describe any negative effects of the trial treatment. The congruity of the ankle joint will be confirmed with radiography. When requested by participants, an experienced orthopedic surgeon will conduct additional ad hoc consultations.
Data collection and management
At the final 2-year follow-up, the participants independently completed questionnaires assessing ankle functional outcome, pain, and quality of life (OMAS, FAOS, VAS, RAND-36) will be collected. The data from the original paper forms will be transferred in duplicate into a secure electronic database protected with access code. If any missing or implausible data are identified while entering data, the research nurses will call the patients to query on them and make a note on the original paper forms. After the completion of all 2-year follow-up visits, these two separate databases are compared for consistency and any discrepancies will be checked against the original paper forms. The resulting ‘master database’ is used in data analysis.
Sample size
The sample size calculation was performed assuming a two-arm study (surgery vs non-operative treatment). In our previous study assessing surgery for unstable ankle fractures with the same primary outcome,35 the mean OMAS score was 79.6 (SD 15.5) at the 2-year follow-up. During the design of the present trial, no estimate for minimal clinically relevant change existed for OMAS. In the absence of better evidence, we organized a focus group discussion among experts to define the appropriate estimate for non-inferiority margin. The panel reached a consensus that a 10% difference in 0–100 OMAS scale would not be clinically significant, which was then used to derive our non-inferiority margin (10% equals eight points in the OMAS scale, Cohen’s d=0.215, indicating a small effect size). With α=0.05, power 80% (1-β=0.8), a non-inferiority margin of 10% (8 points), and with a dropout rate of 20%, the required sample size stood at 63 patients per group (total n=126). The method for sample size calculation is similar to that described in our previous study.28
Sample size calculating formula:
Where: r=1, when equal n per group.
σ2 = estimated population variance (SD 15.5 ≈ 16)
, when α=0.05 and β = 0.20 (power=0.8).
= estimated true mean of treatment A (=80 OMAS points, operative treatment).
= estimated true mean of treatment B (=80 OMAS points, non-operative treatment).
d= non-inferiority marginal (=8 OMAS points).
Statistical analysis
Primary analysis
The trial was primarily designed to ascertain whether non-operative treatment is non-inferior to surgery, 2 years after the injury, with the primary outcome, the OMAS. Only the primary analysis, non-operative treatment versus surgery, will be used to assess non-inferiority. For the primary time point, non-inferiority of non-operative treatment to surgery will be claimed if the lower limit of the confidence interval (for differences in means in OMAS) is greater than −8.0 in the primary comparison. According to the Consolidated Standards of Reporting Trials (CONSORT) statement for non-inferiority and equivalence,24 secondary outcomes can be managed using either a superiority or an equivalence framework. In our trial, all secondary outcomes will be assessed with a superiority hypothesis, but as the trial was not powered for these comparisons, we will merely consider them to be supportive, exploratory, and/or hypothesis generating.
The primary analysis will be performed according to the intention-to-treat principle. In the intention-to-treat analyses, the participants will be included as randomized. The results will be reported following the CONSORT statement.24 36 We will quantify the treatment effect on an intention-to-treat basis as the absolute difference between the groups in the OMAS score (primary outcome) with the associated 95% CIs and p values at 24 months after the randomization (primary time point).
Secondary analyses
We will quantify the treatment effect on an intention to treat basis as the difference between the groups and p values in the secondary outcomes (FAOS, VAS, RAND-36, ROM) where applicable. Categorical variables (occurrence of treatment-related adverse events and non-union) are analyzed with χ2 test or Fisher’s exact test and Wilson’s estimate for the CI of the absolute risk difference.
Adverse events (complications and harms) will be reported descriptively. If the number of events is large enough, an analysis between study arms will be performed.
The baseline characteristics of the participants will be summarized by group, reported as a mean (SD) or median (25th–75th percentiles) for continuous variables and count (per cent) for categorical variables.
Sensitivity analyses
To safeguard against the risk of falsely claiming non-inferiority in case of protocol violations or cross-overs,36 we will also conduct preplanned per-protocol and as-treated analyses (sensitivity analyses). The per-protocol population will be the subset of the intention to treat population who have received the treatment they were randomized to and who did not receive any other treatment, that is, the patients with a treatment conversion will be excluded. In the as-treated analysis, the groups will be analyzed according to their last treatment modality (surgery or non-operative treatment). No other subgroup analyses are planned.
We intend to perform the sensitivity analyses with the same principles as the primary (and secondary) analyses. However, in case of missing data in primary and secondary outcomes we will use multiple imputation to handle missing data. The imputation algorithm, fully conditional specification, uses a specific univariate model for each variable and, for each specific imputed dataset, iteratively imputes each variable with missing values and uses the imputed values in the imputation of other variables. Also, in case of unbalance in baseline data, adjusted linear regression model will be used when applicable.
The data will be analyzed using IBM SPSS Statistics (V.23 or higher, IBM).
Blinded data interpretation
To safeguard against biased interpretation of the trial data, we will interpret the results of the trial according to a blinded data interpretation scheme,37 a procedure we have found helpful in our previous trials.38–40 Briefly, an independent statistician provides the Writing Committee with blinded results from the preliminary analyses, the groups labeled as group A and group B. The Writing Committee then considers the interpretation of the results until a consensus is reached and agrees in writing on the alternative interpretations of the findings. Once a consensus is reached, the minutes of this meeting will be signed by all members of the Writing Committee. Only after this common agreement is reached, the data manager will break the randomization code and the correct interpretation is chosen.
Monitoring
Data monitoring
We will conduct the study without a data monitoring committee (DMC). Both treatment methods are widely used in daily practice and there is prior evidence that both trial interventions provide acceptable results.22 Since there is no DMC, we will not conduct an interim analysis during the trial.
Protocol amendments
All modifications of the study protocol will be communicated by updating the ClinicalTrials.gov trial registration.
Access to data
The research assistant in the study hospital is the only person who has access to the electronic trial data during the data collection. After a ‘master dataset’ is formed from the primary data, access to the dataset will be limited to trial statistician. The codes of the two treatment groups will be known only by the research assistant until the blinded data interpretation has taken place.
Dissemination policy
The findings of this study will be disseminated through peer-reviewed publications and conference presentations. Trial patients will be sent an information leaflet summarizing the results after the primary analysis is published.
Discussion
Surgery represents the mainstay in the treatment of unstable fibula fractures but there is emerging evidence to question the prevailing practice. We designed our SF trial as a non-inferiority trial. According to this rationale, we set out to assess whether the outcome of non-operative treatment is sufficiently close to that of surgery with no excess harms. Non-inferiority of the new treatment with respect to the reference treatment is of interest on the premise that the new treatment has some other advantage, such as greater availability, reduced cost, less invasiveness, fewer adverse effects (harms) or greater ease of administration.24
Our rational for choosing OMAS as the primary outcome measure and 24 months as the primary time point are as follows: We consider (recovery of) ankle function the most relevant outcome to ankle fracture patients. To date, OMAS is the only validated assessment tool for ankle function in patients with an ankle fracture.23 29 Although we acknowledge that some may feel strongly for the primacy of rapid return to normal daily activities and work, we remind that 2 years is commonly considered a gold-standard follow-up time in fracture trials and it has the advantage of capturing majority of harms related to both treatments. Having said that, according to existing understanding, the development of post-traumatic osteoarthritis after an ankle fracture may take a far longer period of time than 2 years.41 42 Therefore, we plan to extend the follow-up of these patients beyond the primary 2-year follow-up.
Because proof of exact equivalence is impossible, a critical methodological decision specifically related to non-inferiority design is the appropriateness of the margin of non-inferiority (Δ): How much worse can the outcome of a new treatment be in relation to the reference treatment to still consider the difference acceptable (clinically irrelevant)? According to the CONSORT guidance on non-inferiority trials,24 the margin of non-inferiority (Δ) should be specified and preferably justified on clinical grounds, as too large a Δ will increase the risk of accepting a truly inferior treatment as non-inferior. A recent study reported that the smallest real difference (SRD) for the OMAS score is 12 points,28 which is higher than our Δ (eight points). Although SRD cannot be directly translated to the margin of non-inferiority as it depicts the ‘smallest detectable change’ for a single subject, while the margin of non-inferiority should represent the smallest change that indicates a real (clinical) improvement or worsening for a group of subjects, we feel that our Δ is reasonable.